- Visibility 17 Views
- Downloads 2 Downloads
- DOI 10.18231/j.ijpca.2021.021
-
CrossMark

Analytical tools in protein structure determination
Introduction
Proteins are one of the most vital biomolecules required for all biological function in our body. The paramount importance of protein can be understood by its considerable role in formation of genetic materials of the living organisms.[1] Proteins are made up of a series of amino acids coupled by the peptide bonds. Nearly 20 types of amino acids are known to contribute to the formation of proteins.[2] The amino acids are linked by condensation of carboxyl group of one amino acid to the amine group of other amino acid. If 8-10 amino acids are joined in a sequence, they are called as oligopeptides, whereas chain containing more than 50 amino acids are known as polypeptides.[3] The functional properties of protein can only be understood by exploring the unique structure adopted by protein as both are interrelated.[4]
The structure of protein can be studied in four stages ([Figure 1]) namely primary, secondary, tertiary and quaternary. The number and sequence of amino acids can be characterized by the primary structure of the protein. Each chain has unique sets of amino acid which is formed by the gene signaling.[5] Interaction between closer segments of amino acid chain gives rise to the secondary structure of protein. Two types of structures may be formed as a result of these interactions, namely α-helices and β-pleated sheet. In α-helices, there is formation of hydrogen bond between C=O group of one amino acid and N-H group of another amino acid that is four down the chain giving rise to a helical structure, while the R groups of the amino acids interact freely as they stick outward from the α-helix. Whereas, in β-pleated sheet, there is hydrogen bond formation between two or more segments of a polypeptide chain giving rise to sheet-like structure and the R groups are present above and below the plane of sheet. Most of the proteincontain both α-helices and β-pleated sheets, while some may contain just one type or do not form either type of secondary structure.[6], [7] Three-dimensional orientation of protein molecules is called tertiary structure. Interactions between the R groups of amino acids makes up the tertiary structure. These interactions include non-covalent forces like Vander Waals interaction, hydrophobic interactions, electrostatic bonds. For example, R groups with like charges repel each other, while those with opposite charges can form an ionic bond. Similarly, polar R groups can form hydrogen bonds and other dipole-dipole interactions. Hydrophobic interactions also contribute to the tertiary structure in which nonpolar, hydrophobic R groups cluster together on the inside of the protein, leaving hydrophilic amino acids on the outside to interact with surrounding water molecules. Disulphide bonds are also important as covalent linkages between the sulphur-containing side chains of cysteines, are much stronger than the other types of bonds that contribute to tertiary structure.The steric correlation between amino acids closes enough in three-dimensional folding is clearly depicted by the tertiary structure[8], [9] When several polypeptide chains come together, they form quaternary structure. Each polypeptide chain contributing to quaternary structure of protein is known as a subunit. Non-covalent interactions along with disulfide bonds lead to stable state of quaternary protein structure.[10] Proteins acquire native confirmation (most stable) by natural automatic folding. Some proteins fold independently while some utilize molecular chaperons. Predominantly, the tertiary structure of protein is the most stable out of all four structures mentioned above. [2], [3]

The intricacy of the protein’s structures makes its determination very arduous even with the use of latest advanced technology. It is a well-known fact that structure and function of protein are profoundly inter-related. Recognition, interaction and action power of proteins highly depend on their conformations and bonds. Modification in any region of protein structure might affect its function. Therefore, determination of protein structure is extremely important for gaining insights into various substantial biochemical aspects of living organisms.
Structure determination of protein
Sanger[11] was the first person to sequence protein insulin in 1955. Structure prediction begins at one-dimensional model of target protein and terminates at three-dimensional model. Protein structure determination involve multi-step process.[4] All the protein samples need to be investigated are purified using different chromatographic techniques.[12] Column chromatography is most widely used technique for fractionating proteins in a mixture. Stationary phase containing solid matrix with multiple microbeads is used for better separation of the sample. The difference in affinity of sample for mobile phase and stationary phase causes different flow rates and thus varying result are obtained. After separation the chromatogram can be visualized by ultraviolet detector and then fractionated portion are collected in tubes by fraction collector.[12] Currently advanced version of column chromatography that is High Performance Liquid Chromatography (HPLC) is being used. It involves passage of mobile phase with a high pressure for faster separation of the sample.[13] Proteins being charged moleculehave the ability to interact with the oppositely charged particles. This property of protein is advantageously used in Ion-exchange chromatography. Electrostatic attraction between the stationary phase (charged immobilized solid matrix) and protein sample is used for its separation.[14] Likewise, reverse chromatographic technique can be used for the non-polar moiety of the protein. Non-polar molecule may be embedded in the stationary phase matrix and the weak interaction between the latter and the non-polar protein residues helps in separation of the sample. The amount and orientation of non-polar amino acid guides the separation in reverse chromatography.[15] Another technique named gel permeation chromatography fractionates molecules on the basis of their size. The gel used has beads with pores which help in the penetration of small molecules only. Two compartments containing aqueous solutions are used, internal compartment is situated inside the bead (accessible by small molecules) and external is situated outside the bead (accessible by both small and large molecules). [16] Affinity chromatography is also employed sometimes for purification of proteins. This method depends on the inclination of protein molecules towards specific ligands according to their individual characteristics. Affinity beads suspended in an aqueous buffer are formed by attachment of the small beads to the ligand. These affinity beads are added to column and the sample solution is then passed through the column. According to the fondness between sample protein and ligand the attachment takes place. Separated proteins can be recovered when beads are washed with buffer solution at lower pH.[12] Recombinant DNA technology is also being used to develop DNA that encodes for particular protein. This technique is a modification of the affinity chromatography and is immensely useful.[17] At isoelectric point, proteins exist as zwitter ions, this property is used in protein electrophoresis techniques for separation. A polyacrylamide gel saturated with buffer solution is used as a migration medium, charged proteins migrate under the influence of electric field according to the nature of their charge. The electrodes are connected to power source at one end of gel (to produce electric field) and the protein sample is applied at the other end. The migration rate is directly proportional to the net charge on protein and inversely proportional to the size. By altering the pH, isoelectric point can be reached. At this point, the proteins cannot migrate due to net charge zero. In this way different proteins are obtained and one clear band is observed in case of pure proteins. Protein electrophoresis may be regarded as an effective way for fractionation of protein.[17], [12], [13], [14], [15], [16], [18]
Primary structure of protein
The first step in determining the primary structure of protein is to identify which amino acids are present and the molar ratio of each. A purified mass of protein is subjected to acid or alkaline hydrolysis to get individual amino acids. The amino acids are separated using chromatography and molar ratio of each amino acid is determined used amino acid analyzer. Next, the disulfides bridges are broken down using oxidation followed by peptide mapping which involve enzymatic break down of protein into smaller fragments. Finally, the proteins can be sequenced using Edman degradation method or Mass spectroscopy.[19], [20]
Edman degradation is one of the most widely used method for determination of primary structure of protein. In 1950, this method was proposed by Pehr Edman and is carried out manually. Briefly, a sequence of chemical reactions is used to cleave the amino acid residue present at the N-terminus of polypeptide chain. This extraction results in exposing of another amino acid residue with free α-amino group. Reduplication of same reactions yields all the amino acid residues one by one. Eventually, all the residues are analyzed and structure is predicted. Thus, this method involves cleavage, separation and identification of one amino acid at a time from amino end of a peptide.[20], [21] In 1967, Edman and Begg automated this method using an instrument known as sequenator.[22] Further advancement was use of solid phase-based method by Laursen in which the chains were covalently bound to the solid matrix. The method had an advantage of reduced loss of small residues during degradation process as they got stuck in the matrix.[23] Hewicket al. in 1981 devised modification of sequenat or known as gas phase sequenator.[24] Now a days, Edman degradation is carried out automatically using overlap method to determine amino acid sequence. Specific enzymes are used for chopping the proteins into fragments. For example, trypsin cleaves at arginine and lysine residues leading to formation of numerous fragments. By tracing the two overlapping fragments from the cleaved sequence the order of chain can be determined.[25] Although, Edman degradation serves a significant purpose but it is a long-winded process and has some shortcomings. It targets the amino acid residues on N-terminus which is hindered in case the amino acid residue is modified. Also, if the peptide is obstructed at the N-terminus then the Edman degradation cannot be carried out.
A modern approach which can surmount all these disadvantages is Mass spectrometry (MS) in which the charged fragments get separated under the influence of electric and magnetic field in vacuum based on their mass to charge ratio. It is a rapid technique which helps in fractionating fragments within seconds, whether the protein is pure or partially pure. Briefly, the purified protein is subjected to gel electrophoresis and the bands containing fragments are cut down. The fragments are dissolved using chemicals or enzymes and the obtained peptide fragments are separated by chromatography followed by MS.[26] Various modified technologies are available for ionization of sample for e.g., ElectrosprayIonization (ES) and Matrix-Assisted Laser Desorption/Ionization (MALDI). ES method has several advantages such as fragile molecule can be easily ionized, proteins with higher mass can be separated and at times the non-covalent interactions can be maintained. In MALDI, solid matrix is used for carrying sampleand laser lights are used for ionization. Moreover, it is uninfluenced by impurities, buffers and additives with rapid activity.MS is highly sensitive therefore a minute quantity of protein sample is required for the sample analysis.[27] Fourier Transform Ion Cyclotron Resonance (FT-ICR) and Time Of Flight (TOF) are employed in conducting whole protein mass analysis.[28], [29] The MALDI-TOF instrument is most frequently used for mass analysis of proteins as they help in attaining peptide mass fingerprints.[30] Additionally, Quadrupole Ion Trap and Multiple Stage Quadrupole-Time-Of-Flight methods are also employed.[31] Tandem mass spectroscopy can be used to sequence to 20 to 30 amino acids in length. This method involves ionization of polypeptide and the first mass spectrometer separate these charged peptides by their mass-to -charge ratio. These peptides then enter a collision chamber and split into smaller fragment which are then measured at second mass spectrometer. The resultant spectrum can determine the peptide sequence by differences in mass of the fragments. Accuracy and speed for fragmentation can also be obtained by Tandem mass spectrometry.[32] Presently two approaches are used for mass fragmentation: Top-down and Bottom-up. Top-down approach uses whole protein mass for ionization which makes it difficult to characterize due to complexity of whole protein mass. In contrast, bottom-up approach uses fractioned mass for ionization hence, it is more efficient and accurate as characterization takes place at peptide level due to initial fragmentation.[33] Protein identification by MS takes place either by peptide mass fingerprinting method or de novo sequencing. In case of peptide mass fingerprinting, database search is performed to compare and find match of the sample protein fragments.[34] De novo sequencing uses mass comparison of known proteins and sample protein.[35] Three-dimensional structures of proteins can be investigated in various ways using MS for e.g., Hydrogen-deuterium exchange type MS has been used since a long time especially for proteins with complex structure.[36] Proteins post-translational modifications, associated subunits, interactions and proteomics studies are also possible by this explosive method.[26] In short, we can say that MS serves in multidimensional ways in the case of protein structure determination.
Secondary structure of protein
Generally, two methods are used to characterize the secondary structure and stability properties of protein namely, Circular Dichroism (CD) and Fourier-Transform Infrared Spectroscopy (FT-IR).
Circular Dichroism (CD) is an unequal absorption of left-handed and right-handed circularly polarized light. When this circularly polarized light passes through an asymmetric or chiral environment (as present in proteins) illumination can be observed. CDis based on the excitation of electronic transition in amide groups due to exciton interactions. This result in different types of secondary structures with differential absorption in the far-ultraviolet region of the spectrum (180-250 nm). This helps in deciphering the protein backbone as well as side chains.[6], [7]
Fourier-Transform Infrared Spectroscopy (FT-IR)is based on the vibrations of the molecules in the sample. Generally, IR spectrum of protein displays nine amide bands due to vibrations of protein backbone a well as amino acid side chains. Absorptions caused due to C=O bond stretching are termed as amide I and the one due to N-H bond bending are termed as amide II. As N-H and C=O bonds are responsible for the formation of hydrogen bonds the locatio ns of these amide bonds are very important for finding out secondary structure conformation of protein. In comparison to the intrinsic width of the band, the shifts in amide I bands are usually lower. This leads to formation of a broader peak in place of chain of resolved peaks for specific type of secondary protein. Second derivatives, band-curve fitting and fourier self-deconvolution are the mathematical tools that help in resolving the peaks overlapping peaks.[37], [38], [39] Overall, FT-IR has been a sensitive and versatile method for secondary protein structure determination.
Tertiary structure of protein
Proteins try to fold themselves to attain the most stable conformation which is termed as its native state. The Gibbs free energy is very low for folded conformation compared to the unfolded one. Various protein conformations carry approximately similar energy therefore the proteins constantly keep wavering between these conformations.[40] Currently X-ray crystallography and Nuclear Magnetic Resonance (NMR)spectroscopyare the most commonly used techniques for tertiary structure elucidation. Apart from that cryogenic Electron Microscopy (cryo-EM) and Dual Polarization Interferometry (DPI) techniques are also being used.
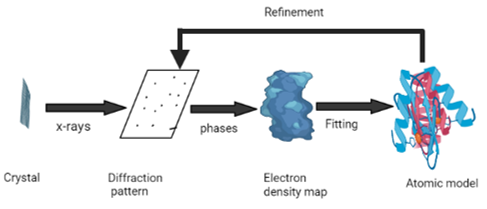
X-ray crystallography undoubtedly remains one of the most used method for rigid protein structure determination ([Figure 2]). Protein Data Bank (PDB) is a unique storehouse of three-dimensional protein structure, nucleic acids and their complexes with small molecules. Nearly ninety percent of PDB structures have been investigated through X-Ray crystallography.[41] Once the protein is purified, the next step is to form crystals of sufficient quality to collect high-resolution data for structure determination. An intense X-ray beam is exposed to protein crystal. The atoms present in protein diffract the X-ray beam to form a diffraction pattern in the form of spots. These spots are analyzed later for detection of structure.[42] The process of crystallization is generally slow, resource-intensive step with a low success rate which is attributed to poor protein quality. Therefore, it is necessary to assess the quality of a protein as well as optimize conditions such as pH, temperature, salt and protein concentrations, and cofactors before performing experiments. Crystallization is also done using automated technologies using small protein sample volumes and also increases the efficiency for the screening crystallization conditions.[43] Now a days, experiments are carried out in parallel using the second-generation crystallization robots, utilizing nanoliter-sized drops of protein sample to screen for the optimal conditions. Additionally, they are equipped with inbuilt image capture and analysis systems, which monitor the drops and look for the appearance of crystals using edge-detection methods.[44] Detectors used are single-photon counters, photographic film, image plates and area detectors. Another rate-determining stepis the manual intervention required for the crystal mounting and alignment in the X-ray beam.[42] A novel method for automatic mounting, optical crystal alignment, and the data collection has been reported recently. It exhibits the same range of accuracy as the manual process and allows the data collection using conventional X-ray systems. In addition, the number of high-brilliance synchrotron X-ray beam lines dedicated to macromolecular crystallography has been increased significantly, and thus data collection time at these facilities are dramatically reduced.[44], [45] The major drawback of X-ray crystallography lies in the inability to probe the flexible protein conformations which are not able to form crystals.[42]
Nuclear Magnetic Resonance (NMR) spectroscopy has proved to be an outstanding technique not only for investigating protein structure ([Figure 3]) but also to determine its functional dynamics for better understanding of macromolecules. Fortunately, this technique doesn’t degrade the sample during the experiment which builds a trust of obtaining the accurate information regarding the protein structure. For NMR, solution form of sample is required whereas for X-ray crystallography crystalline forms are mandatory. However, the determination of structure by NMR is currently limiteddue to size constraints and the lengthy data collection and analysis time. Nevertheless, the NMR spectroscopy still plays the significant role in the structural proteomics even with its current limitation. Proteins containing amino acid residues 100 or less have been rewardingly characterized by NMR spectroscopy. Briefly, a suitable sample containing 1 mM protein in 500 mL is prepared and subjected to radiofrequency waves and external magnetic field. For protein samples with molecular mass more than 10 kDa, enhancement with 13C and 15N isotopes is required to resolve spectral overlap in proton NMR. The NMR spectra obtained after suitable data processing gives information about the protons present in the sample. Variation in the chemical shift due to varying conformations plays a very important role in deriving the structure.[46] In addition, the sensitivity of the acquired NMR data relies strongly on the performance of the NMR probe, a sophisticated electronic device used to detect NMR signals.[47] Finally, the NMR structure can be refined using the conformational energy force fields. Correlation spectroscopy (COSY), a type of two-dimensional spectroscopy, is used for homonuclear NMR spectroscopy. Different modification of COSY like Total correlation spectroscopy (TOSCY) and Nuclear over hauser effect spectroscopy (NOESY) are also used extensively. The transient magnetization technique is used in these methods to determine the interactions between different atoms in a protein. Especially NOESY serves to depict the proton pairs as neighbors in the form of graph even if they are far apart in the primary structure[48], [49] 2-D and 3-D NMR are preferred over 1-D NMR due to occurrence of multiple overlapping lines in 1-D NMR.

As we know, the major challenge in NMR spectroscopy is the reduction of data collection time required for the structure determination. Deuteration of protons provides samples with improved signal-to-noise ratio results in sharper line widths and longer transverse relaxation time.[50] NMR has largely been able to characterize only the structure of small proteins due to excessive overlapping peaks obtained in case of large proteins. Moreover, there is less time for signal detection due to rapid relaxation of magnetization for large proteins. Transverse relaxation optimized spectroscopy (TROSY) based on selection of the slowly relaxing NMR transitions provides significant sensitivity enhancement for the large proteins.[51] Recently, various computerized techniques are being used for automated structure determination from the NMR spectra such as UNIO AND FLYA.[52] Another approach is NOESY-Jigsaw, in which sparse and unassigned NMR data are used reasonably and accuratelytoalign and assess secondary structure. The information obtained is thus useful to predicts and analyze folds before full structure determination.[53] In addition, complete automation with visual verification capacity has also been possible by using APES, PONDEROSA [54] etc. Another important parameter needs to develop involves automated analysis of NMR data. The various design of cryogenic probes will definitely assist in reducing thetime required for data collection in the NMR-based structural proteomics. Some automated programs like NOAH, AUTOASSIGN, ARIA, ANSIG, TATAPRO, and SANE, hold great promise in reducing for the amount of time and automated analysis of NMR assignments and 3-D structures of the proteins up to 200 amino acids.[55] These technologies together provide the basis for high-throughput NMR, and they are particularly valuable for samples with limited stabilities and low solubilities.
X-ray crystallography and NMR spectroscopy are indeed the best techniques benefitting the scientists to decipher the structure and conformations of protein even at the atomic level. But the incapability of X-ray crystallography to detect the structure of flexible proteins and inability of NMR technique to investigate large proteins have been the key obstacle in elucidating protein structure perfectly. An advanced technique known as cryo-electron microscopy (cryo-EM) has been successful in subduing these problems. Cryo-EM is used for the protein structure determination and biological macromolecular assemblies. Electron microscopic technique provides sufficient resolution to produce atomic models of the proteins. Specifically, the single- particle cryo-EM has been proved to be a boon in analyzing heterogenous,large and dynamic conformations effectively with resolution less than nanometer. Computational averaging of numerous two-dimensional projection images is used to form three dimensional conformations of non-crystalline samples using single-particle cryo-EM.[56] Like the purity of protein and crystal formation were chief concern in obtaining a good diffraction pattern in X-ray crystallography, similarly here the sample purity plays an important role in obtaining perfect data. Therefore, if the sample is heterogenous then it needs to be made homogenous before performing the experiment. Although gel-permeation chromatography is used for partitioning and decreasing heterogeneity but the outcome is not very suitable. Advantageously negative staining method serves as a good option for rapidly optimizing the heterogenous sample formation. In negative staining, the sample is attached to the EM grid and exposed to high electron density. Due to difference in electron density a high contrast is obtained. Where the density of particle is less, the electrons are scattered at lower pace making it visibly darker and vice-versa. Negative staining cannot provide very good resolution but is of paramount importance in preparing the sample for heterogenous kind of particle analysis[57], [58] cryo-EM has also been reported to avoid dehydration of sample after being placed in vacuum of electron microscope. Native structure is well maintained using flash-freezing method. Its usefulness can be seen when high resolution images are obtained by this freezing technique.[59] Two-dimensional micrograph processing is most time-consuming step, regulating the electron beam trajectory is one effective way to overcome this problem, although some comatic aberrations may develop. Collection of images using phase plates has drawn interests owing to its property of improving image contrast. For example -volta phase plate has produced high resolution structures of some protein samples. Recently, high-intensity laser technology is being used to form new phase plates for better resolution. In single particle cryo-EM, the three-dimensional structures are made by joining pieces of two-dimensional micrographs and image transformation is based on the projection-slice theorem.[60] Conclusively, we can say that cryo-EM is a revolution in the pathway of probing protein structure and has helped extensively.
Dual polarization interferometry (DPI) is anovel advancement in science which provides real-time characterization of protein structure by monitoring the crystal formation. It uses electromagnetic evanescent laser beam wave for probing the molecular layer adsorbed to the waveguide surface. Protein adsorption studies at solid-liquid interface as well as measurement of refractive index and thickness of the film can be done using this method. It is based on the interactions between small molecules and proteins aided with biosensors. Protein-ligand interaction knowledge can also be gained by using DPI.[61]
Quaternary structure of protein
X-ray crystallography is also commonly used for quaternary structure determination. Targeted cross-linking mass spectroscopy (TX-MS) has also found application in determination of quaternary structure of protein. Chemical cross-linking forms the basis of this method. MS helps in developing signals for targeted cross-linked peptides which are studied using computerized techniques.[62] Recently, Forster resonance energy transfer (FRET)technique has shown beneficial results in detecting the protein quaternary structure. FRET is based on the non-radiative energy transfer between excited donor molecule and unexcited acceptor molecule. Donor quenching, increased emission of the donor and decreased time of donor life act as pillars for probing FRET-based imaging. Alternations in conformations and interaction between molecules of proteins involved in movement and adhesion of cells was analyzed by Parsons et al using FRET technique. G protein coupled receptors oligomerization was investigated using time-resolved FRET by Albizu and co-workers. Fortunately, FRET can be used for size and shape determination of protein samples effectively.[63]
ROSETTA is a computational tool for determination of the protein structures. The protein structure is extracted from the PDB, which consist of information of each coordinate of every atom in a protein. Conventionally, the FASTA file (stores one letter code for every amino acid in the sequence) is used for representation of primary structure of protein. BLAST (Basic local alignment search tool) is used for aligning the FASTA file to all possible known sequences to find the common sequences present in the protein. BLAST is a beneficial tool which does comparison between target protein and all proteins present in the database for detecting the common amino acids present in the sample. This helps in finding out the structure of unknown protein with the use of known protein. Subsequent to BLAST data generation, various programs for secondary structure prediction are run to configure the sequences belonging to alpha-helix or beta strand. For example-JUFO, PSIPRED (both use artificial neural networks) and SAM (uses Hidden Markov Models) are used for secondary structure predictions. Next step is to generate fragments. The program repeats every overlapping three and nine residue stretch of the chain of interest, then finds similar stretches of chain resulting in fragmentation, from the proteins having structures determined experimentally, and selects two hundred such fragments structures for every location in the chain. Further fragment database is formed by storing these fragments in the form of a file. Finally, rosetta program is run for prediction of fifty-thousand structural model for individual proteins in the benchmark set. Root mean square deviation (RMSD) values are used for testing the structural models produced by rosetta method. The closeness of a produced structure to its native structure can be determined by its RMSD value.[64], [65]
Conclusion
The eventual goal of protein structure determination is to know about the function of protein as both are interrelated. Protein structure determination methods play an important role in the generation of full structural proteome maps, but many challenges remain. Now a days, experimental methods are coupled with computational methods to determine protein structure fast and accurately. X-ray crystallography is the most common methodology applied to the structural genomics by automating nearly every stage of the process. NMR methods are used for obtaining structural information on macromolecules and molecular complexes. The accurate models generated using Rosetta correlate best with the complexity of the sequence length and the protein topology. Electron microscopy combined with cryo-technology aids in producing high resolution images of biological specimens that provides detail information about their assemblies. Although above-mentioned methods have advantages and disadvantages, the information generated by them is used collectively to elucidate the protein structure.
Source of Funding
None.
Conflict of Interest
None.
References
- Hartleyh. Origin of the word ‘protein’. Nature 1951. [Google Scholar]
- C Ollivaux, D Soyez, J Y Toullec. Biogenesis of D-amino acid containing peptides/proteins: where, when and how?. J Pept Sci 2014. [Google Scholar] [Crossref]
- F Sanger, E O P Thompson. The amino acids sequence in the glycyl chain of insulin. 2. The investigation of peptides from enzymic hydrolysates. Biochemical J 1953. [Google Scholar] [Crossref]
- J C Whisstock, A M Lesk. Prediction of protein function from protein sequence and structure. Q Rev Biophys 2003. [Google Scholar] [Crossref]
- E Kennedy, Z Dong, C Tennant, G Timp. Reading the primary structure of a protein with 0.07 nm 3 resolution using a subnanometre-diameter pore. Nat nanotechnol 2016. [Google Scholar] [Crossref]
- S Y Venyaminov, J T Yang. Determination of protein secondary structure. InCircular dichroism and the conformational analysis of biomolecules. . 1996. [Google Scholar]
- K Kuwajima, H Yamaya, S Miwa, S Sugai, T Nagamura. Rapid formation of secondary structure framework in protein folding studied by stopped-flow circular dichroism. FEBS letters 1987. [Google Scholar] [Crossref]
- I D Kuntz, G M Crippen, P A Kollman, D Kimelman. Calculation of protein tertiary structure. J Molecular Biol 1976. [Google Scholar] [Crossref]
- J Meiler, D Baker. Coupled prediction of protein secondary and tertiary structure. Proc National Acad Sci 2003. [Google Scholar] [Crossref]
- J Janin, R P Bahadur, P Chakrabarti. Protein-protein interaction and quaternary structure. Q Rev Biophys 2008. [Google Scholar] [Crossref]
- A O Stretton. The first sequence: Fred Sanger and insulin. Genetics 2002. [Google Scholar] [Crossref]
- P Cuatrecasas. Protein purification by affinity chromatography: derivatizations of agarose and polyacrylamide beads. J Biol Chem 1970. [Google Scholar]
- C T Mant, Y Chen, Z Yan, T V Popa, J M Kovacs, J B Mills. HPLC analysis and purification of peptides. InPeptide Characterization and Application Protocols. Methods Mol Biol 2007. [Google Scholar] [Crossref]
- H Yang, M Bitzer, M R Etzel. Analysis of protein purification using ion-exchange membranes. Ind Eng Chem Res 1999. [Google Scholar]
- C Megías, J Pedroche, D M Yust, M Alaiz, M G Calle, J Millán. Purification of angiotensin converting enzyme inhibitory peptides from sunflower protein hydrolysates by reverse-phase chromatography following affinity purification. J Agric Food Chem 2009. [Google Scholar] [Crossref]
- M E Pugh, E Schultz. Assessment of the purification of a protein by ion exchange and gel permeation chromatography. Biochem Mol Biol Educ 2002. [Google Scholar]
- W W Ward, G Swiatek. Protein purification. Curr Anal Chem 2009. [Google Scholar]
- J Doly, F Petek. Polyacrylamide gradient electrophoresis for protein purification on the milligram scale. J Chromatography A 1977. [Google Scholar] [Crossref]
- A A Gooley, K Ou, J Russell, M R Wilkins, J C Sanchez, D Hochstrasser. A role for Edman degradation in proteome studies. Electrophoresis 1997. [Google Scholar] [Crossref]
- J M Bailey. Chemical methods of protein sequence analysis. J ChromatographyA 1995. [Google Scholar] [Crossref]
- P Klemm. Manual edman degradation of proteins and peptides. Methods Mol Biol 1984. [Google Scholar] [Crossref]
- P Edman, G Begg. A protein sequenator. Eur J Biochem 1967. [Google Scholar]
- R A Laursen. Solid-Phase Edman Degradation: An Automatic Peptide Sequencer. Eur J Biochem 1971. [Google Scholar] [Crossref]
- R M Hewick, M W Hunkapiller, L E Hood, W J Dreyer. High sensitivity sequencing with a gas-phase sequenator. Methods Enzymol 1981. [Google Scholar] [Crossref]
- H D Niall. Automated edmandegradation:The protein sequenator. Methods enzymology 1973. [Google Scholar] [Crossref]
- M Palmblad, M Ramström, C G Bailey, S L M Maloney, J Bergquist, L C Zeller. Protein identification by liquid chromatography-mass spectrometry using retention time prediction. J Chromatography B 2004. [Google Scholar]
- A Dell, H R Morris. Glycoprotein structure determination by mass spectrometry. Science 2001. [Google Scholar] [Crossref]
- T Tucholski, Y Ge. Fourier-transform ion cyclotron resonance mass spectrometry for characterizing proteoforms. Mass Spectrom Rev 2020. [Google Scholar]
- O A Mirgorodskaya, A A Shevchenko, I V Chernushevich, A F Dodonov, A Miroshnikov. Electrospray-ionization time-of-flight mass spectrometry in protein chemistry. Anal Chem 1994. [Google Scholar] [Crossref]
- L Signor, E B Erba. Matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometric analysis of intact proteins larger than 100 kDa. J Vis Exp 2013. [Google Scholar] [Crossref]
- A Barnes, J Bale, C Constantinidou, P Ashton, A Jones, J Pritchard. Determining protein identity from sieve element sap in Ricinuscommunis L. by quadrupole time of flight (Q-TOF) mass spectrometry. J Exp Bot 2004. [Google Scholar] [Crossref]
- K Biemann, H A Scoble. Characterization by tandem mass spectrometry of structural modifications in proteins. Science 1987. [Google Scholar] [Crossref]
- N L Kelleher, L H Y Valaskovic, G A Aaserud, D J Fridriksson, E K Mclafferty, F W. Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J Am Chem Soc 1999. [Google Scholar] [Crossref]
- D J Pappin, P Hojrup, A J Bleasby. Rapid identification of proteins by peptide-mass fingerprinting. Curr Biol 1993. [Google Scholar] [Crossref]
- N Bandeira, V Pham, P Pevzner, D Arnott, J R Lill. Automated de novo protein sequencing of monoclonal antibodies. Nat Biotechnol 2008. [Google Scholar] [Crossref]
- I Oganesyan, C Lento, D J Wilson. Contemporary hydrogen deuterium exchange mass spectrometry. Methods 2018. [Google Scholar] [Crossref]
- A Adochitei, G Drochioiu. Rapid characterization of peptide secondary structure by FT-IR spectroscopy. Rev Roum Chim 2011. [Google Scholar]
- H Fabian, D Naumann. Methods to study protein folding by stopped-flow FT-IR. Methods 2004. [Google Scholar] [Crossref]
- W Jianhua, W Yali, W Zonghe, H Jianchuan. Advances in Application of FT-IR to the Research of Protein Structure. Chem Peking 2004. [Google Scholar]
- A Hvidt, P Westh. Different views on the stability of protein conformations and hydrophobic effects. J Solution Chem 1998. [Google Scholar]
- H M Berman, T N Bhat, P E Bourne, Z Feng, G Gilliland, H Weissig. The Protein Data Bank and the challenge of structural genomics. Nat struct biol 2000. [Google Scholar] [Crossref]
- M S Smyth, J H Martin. x ray crystallography. Mol Pathol 2000. [Google Scholar] [Crossref]
- A Ilari, C Savino. Protein structure determination by x-ray crystallography. Bioinformatics 2008. [Google Scholar] [Crossref]
- T L Blundell, S Patel. High-throughput X-ray crystallography for drug discovery. Curr Opin Pharm 2004. [Google Scholar]
- A Sharff, H Jhoti. High-throughput crystallography to enhance drug discovery. Curr Opin Chem Biol 2003. [Google Scholar] [Crossref]
- A Cavalli, X Salvatella, C M Dobson, M Vendruscolo. Protein structure determination from NMR chemical shifts. Proc National Acad Sci 2007. [Google Scholar] [Crossref]
- M A Danielson, J J Falke. Use of 19F NMR to probe protein structure and conformational changes. Annual review of biophysics and biomolecular structure. Annu Rev Biophys Biomol Struct 1996. [Google Scholar] [Crossref]
- A Bax. Two-dimensional NMR and protein structure. Annu Rev Biochem 1989. [Google Scholar] [Crossref]
- C Mumenthaler, P Güntert, W Braun, K Wüthrich. Automated combined assignment of NOESY spectra and three-dimensional protein structure determination. J Biomol NMR 1997. [Google Scholar] [Crossref]
- V Chevelkov, K Rehbein, A Diehl, B Reif. Ultrahigh resolution in proton solid-state NMR spectroscopy at high levels of deuteration. Angew Chem Int Edition 2006. [Google Scholar] [Crossref]
- K Pervushin. Impact of transverse relaxation optimized spectroscopy (TROSY) on NMR as a technique in structural biology. Q Rev Biophys 2000. [Google Scholar] [Crossref]
- N Kobayashi, Y Hattori, T Nagata, S Shinya, P Güntert, C Kojima. Noise peak filtering in multi-dimensional NMR spectra using convolutional neural networks. Bioinformatics 2018. [Google Scholar] [Crossref]
- C B Kellogg, A Widge, J J Kelley, M J Berardi, J H Bushweller, B R Donald. The NOESY jigsaw: automated protein secondary structure and main-chain assignment from sparse, unassigned NMR data. J Comput Biol 2000. [Google Scholar] [Crossref]
- C Narayan, K Bafna, L D Roux, P K Agarwal, N Douset. Applications of NMR and computational methodologies to study protein dynamics. Arch Biochemi Biophys 2017. [Google Scholar] [Crossref]
- H L Liu, J P Hsu. Recent developments in structural proteomics for protein structure determination. Proteomics 2005. [Google Scholar] [Crossref]
- M Bonomi, R Pellarin, M Vendruscolo. Simultaneous determination of protein structure and dynamics using cryo-electron microscopy. Biophys J 2018. [Google Scholar] [Crossref]
- J R Harris. Negative staining and cryoelectron microscopy. Micros Today 1997. [Google Scholar]
- M Rames, Y Yu, G Ren. Optimized negative staining: a high-throughput protocol for examining small and asymmetric protein structure by electron microscopy. J Vis Exp 2014. [Google Scholar]
- A Doerr. Single-particle cryo-electron microscopy. Nature Methods 2016. [Google Scholar]
- K Murata, M Wolf. Cryo-electron microscopy for structural analysis of dynamic biological macromolecules. Biochim Biophys Acta Gen Subj 2018. [Google Scholar] [Crossref]
- M J Swann, L L Peel, S Carrington, N J Freeman. Dual-polarization interferometry: an analytical technique to measure changes in protein structure in real time, to determine the stoichiometry of binding events, and to differentiate between specific and nonspecific interactions. Anal Biochem 2004. [Google Scholar] [Crossref]
- S Hauri, H Khakzad, L Happonen, J Teleman, J Malmström, L Malmström. Rapid determination of quaternary protein structures in complex biological samples. Nat Commun 2019. [Google Scholar] [Crossref]
- B Schuler, W A Eaton. Protein folding studied by single-molecule FRET. Curr Opin Struct Biol 2008. [Google Scholar] [Crossref]
- C A Rohl, C E Strauss, K M Misura, D Baker. Protein structure prediction using Rosetta. Methods Enzymology 2004. [Google Scholar] [Crossref]
- T A Ramelot, S Raman, A P Kuzin, X R Ma, L C Actontb, J F Hunt. Improving NMR protein structure quality by Rosetta refinement: a molecular replacement study. Proteins 2009. [Google Scholar] [Crossref]